Manual¶
Contents¶
roboraptor is a comprehensive pipeline and set of tools for analyzing Ribo-seq data. This manual explains the stages in our pipeline, how to use the analysis tools and how to modify the pipeline for your specific context.
Assumptions¶
Our pipeline was designed to run in a cluster computing context, with many processing nodes available, and a job submission system like PBS or SGE. Much of this analysis is computationally intensive. We assume that individual nodes will have several GB of memory available for processing.
Translatome construction¶
Ribo-seq experiments are always single-end sequenced. Ribosome protected fragments range from 28-32 nucleotides and hence most experiments involve 50 bp single end reads. Before mapping, we need to get rid of the adapters ligated at the 3’ end of the fragments as part of the library protocol.
Trimming Reads¶
We use trim_galore for trimming. It automates adapter trimming:
$ trim_galore -o <out_dir> -q <min_quality> <input.fq.gz>
| -o out_dir | Output directory |
| -q min_quality | Trim low-quality ends from reads in addition to adapter removal |
Mapping Reads¶
We use STAR to map reads, though other splice-aware aligners can also be used. The first step is to create an index, preferably using a GTF file. If the index step is run without a GTF file (which is optional), STAR will not be splice-aware.
Creating Index¶
$ STAR --runThreadN <threads>\
--runMode genomeGenerate\
--genomeDir <index_out_dir>\
--genomeSAindexNbases <SA_INDEX_Nbases>\
--genomeFastaFiles <input.fasta>\
--sjdbGTFfile <input.gtf>
| --runThreadN | Number of threads to use |
| --runMode | Flag to set for index mode |
| --genomeDir | Directory to write index files to |
| --genomeSAindexNbases | |
| min(14, log2(GenomeLength)/2 - 1), this must be scaled down for small genomes | |
| --genomeFastaFiles | |
| Path to reference fasta | |
| --sjdbGTFfile | Path to GTF file |
Mapping¶
Often Ribo-seq experiments have a matching RNA-seq library. Ideally, the RNA-seq library should be as similar to Ribo-seq library and hence will often be single-ended. We recommend both RNA-seq and Ribo-seq samples be mapped using the following parameters:
$ STAR --runThreadN <threads>\
--genomeDir <input.index>\
--outFilterMismatchNmax 2\
--alignIntronMin <ALIGN_INTRON_Nmin>\
--alignIntronMax <ALIGN_INTRON_Nmax>\
--outFileNamePrefix <params.prefix> --readFilesIn <input.R1>\
--outSAMtype BAM Unsorted\
--readFilesCommand zcat\
--quantMode TranscriptomeSAM\
--outTmpDir /tmp/<params.name>_tmp\
--outReadsUnmapped Fastx\
| --runThreadN | Number of threads to use |
| --genomeDir | Path to index directory |
| --outFilterMismatchNmax | |
| Allow a maximum of mismatches=2 | |
| --alignIntronMin | |
| Minimum intron size. Any genomic gap is considered intron if its length >= alignIntronMin. (Default = 20) | |
| --alignIntronMax | |
| Maximum intron size (Default = 1000000) | |
| --outFileNamePrefix | |
| Prefix for output files | |
| --readFilesIn | Path to input fastq.gz |
| --outSAMtype | Output an unsorted BAM file (outtype=BAM Unsorted) |
| --readFilesCommand | |
| cat/zcat depending on input is fq/fq.gz | |
| --quantMode | Also output BAM aligned to the transcriptome |
| --outTmpDir | Directory to use for writing temporary files |
| --outReadsUnmapped | |
| Write unmapped reads to separate fastq file | |
Sorting and Indexing¶
STAR outputted BAM files are not sorted. We need a BAM file sorted by coordinates.
$ samtools sort <prefix>Aligned.out.bam -o <output.bam> -T <tmpdir>_sort &&\
$ samtools index <prefix>Aligned.out.bam
Additionaly, we also need BAM file sorted by name, since htseq-counts (and featureCounts) prefer a BAM sorted by name in their default mode.
$ samtools sort -on <input.bam> -T <tmpdir> -o <output.bam> &&\
$ samtools index <output.bam>
Translatome analysis¶
Once we have the bams, we are ready for downstream analysis. The downstream step often involves a number of steps. The following list summarises these steps along with their recommended values (wherever applicable):
- Quality Control
- Number of uniquely mapped reads : >=5M
- Periodicity : TODO
- Ratio of CDS/(5’UTR+3’UTR) : >1 after length normalization
- Fragment length distribution : Peak around 28-32 nt
- Metagene analysis
- P-site offsets : Around 12-14 nt upstream of the start codon when counting based on 5’end
Counting uniquely mapped reads¶
The first step is to simply caculate the number of uniquely mapped reads. We recommend a minimum of 5 million reads for any downstream analysis. TODO: list different recommendation for different species
$ riboraptor uniq-mapping-count --bam <input.bam>
–bam input.bam Path to bam file
Read length distribution¶
An ideal Ribo-seq library is expected to have 28-32 nt long fragments most enriched. We can calculate enrichment and plot the fragment size distribution using riboraptor.
Readd length distribution can be calculated using the read-length-dist subcommand:
$ riboraptor read-length-dist --bam <input.bam>
This will print out the read length and associated counts on the console. In order to visualize thhese counts as a barplot, we can use the plot-read-dist subcommand:
$ riboraptor read-length-dist --bam <input.bam>\
| riboraptor plot-read-dist --saveto <output.png>
Metagene Analysis¶
A metagene plot is used as a summary statistic to visualize the distribution of ribosome protected fragments along the positions of a gene often starting (ending) at the start (stop) codon. This is useful for estimating P-site offsets. The ribosome subunuits are known to protect 28-32 nt and hence the P-site is often located 12 nt downstream the 5’ position of the mapped read.
Creating bigWig file¶
To perform metagene analysis, we will work with bigWig format. in order to do that, we need an intermediate bedGraph file. This can be done using bam-to-bedgraph subcommand:
$ riboraptor bam-to-bedgraph --bam <input.bam>
This will print the bedGraph to console. this cna be piped to bedgraph-to-bigwig subcommand:
$ riboraptor bam-to-bedgraph --bam <input.bam> \
| riboraptor bedgraph-to-bigwig --sizes <genome.sizes> --saveto <output.bw>
We now have <output.bw> ready for further downstream analysis.
Distribution in 5’UTR/3’UTR/CDS regions¶
TODO (See Example)
Metagene plot¶
TODO (See Example)
Example¶
We will use two samples from GSE94454 , one RNA-seq sample (SRR5227310) and one Ribo-seq sample (SRR5227306) as examples for examples that follow.
$ riboraptor uniq-mapping-count --bam data/SRR5227310.bam
28637667
This is a pretty deep library.
$ riboraptor read-length-dist --bam data/SRR5227310.bam\
| riboraptor plot-read-dist --saveto SRR5227310.png
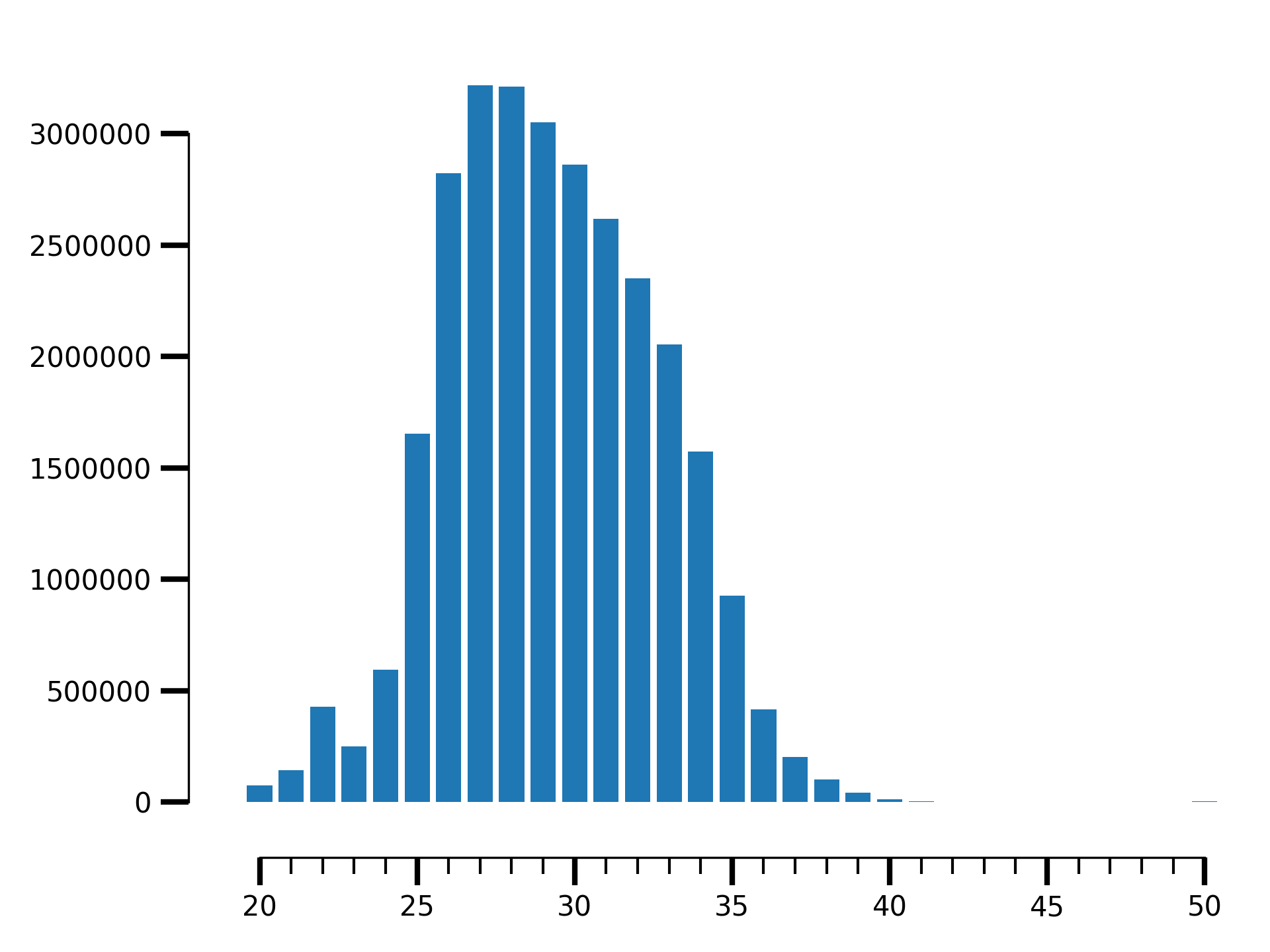
Fragment length distribution for SRR5227310
How enriched is it in 27-32 nt fragment range?
$ riboraptor read-length-dist --bam data/SRR5227310.bam\
| riboraptor read-enrichment
(Enrichment: 1.52768004237, pval: 0.458943823895)
So the fragment length distribution doesn’t seem to be enriched. We next perform metagene analysis. Ribo-seq data is expected to have an inherent periodicity of 3, since ribosomes move one codon at a time during active translation.
$ riboraptor bedgraph-to-bigwig -bg data/SRR5227310.bg -s hg38 -o data/SRR5227310.bw
$ riboraptor metagene-coverage -bw data/SRR5227310.bw \
--region_bed hg38_cds --max-positions 500 \
--prefix data/SRR5227310.metagene --offset 60 --ignore_tx_version
$ riboraptor plot-read-counts \
--counts data/SRR5227310.metagene_metagene_normalized.pickle\
--saveto data/SRR5227310.metagene.png
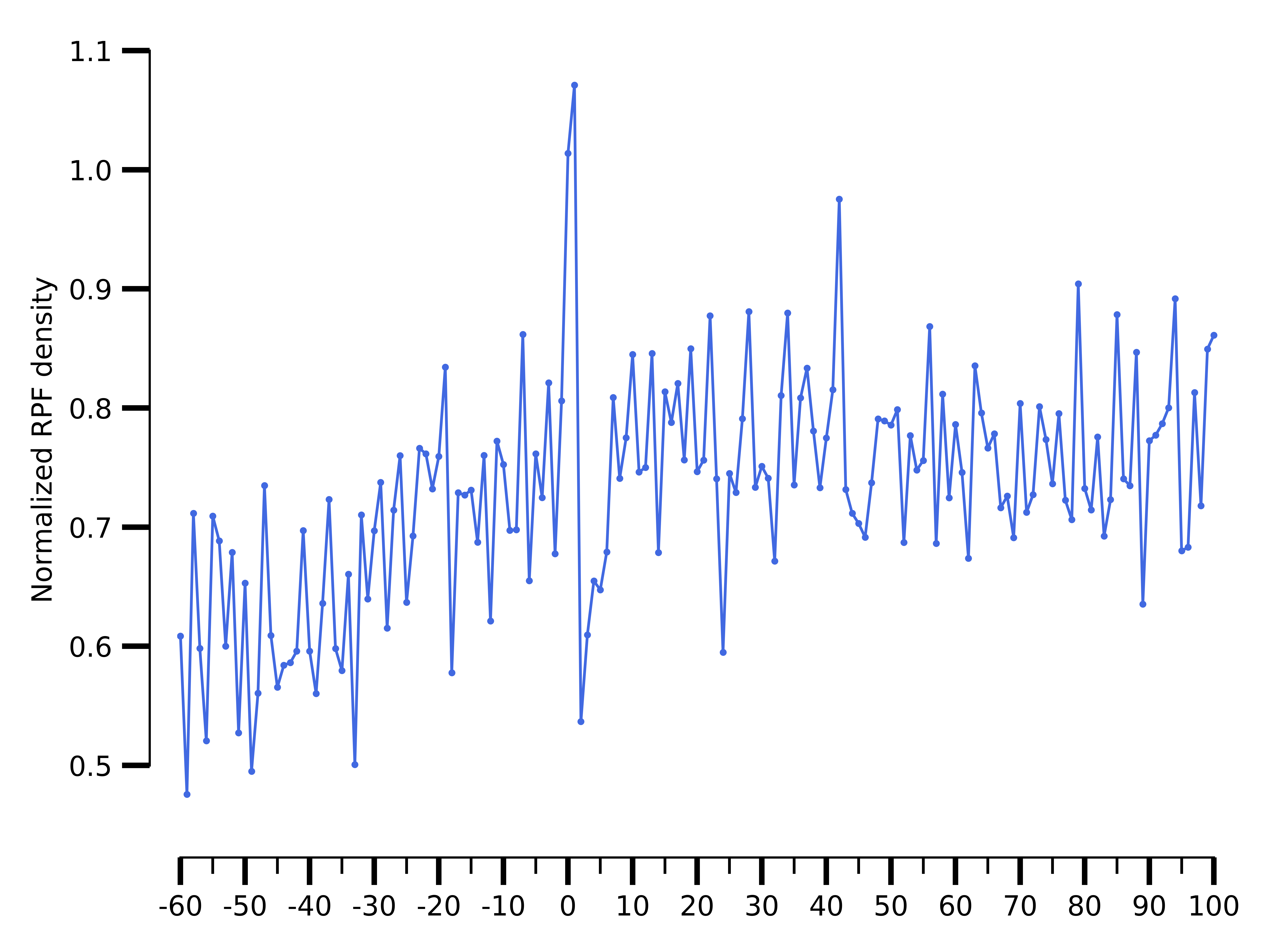
Metagene distribution for SRR5227310
Since metagene gives a summary statistic, we can also look at the abolute counts distribution per frame:
$ riboraptor plot-framewise-counts --counts data/SRR5227310.metagene_metagene_raw.pickle\
--saveto data/SRR5227310.framewise.png
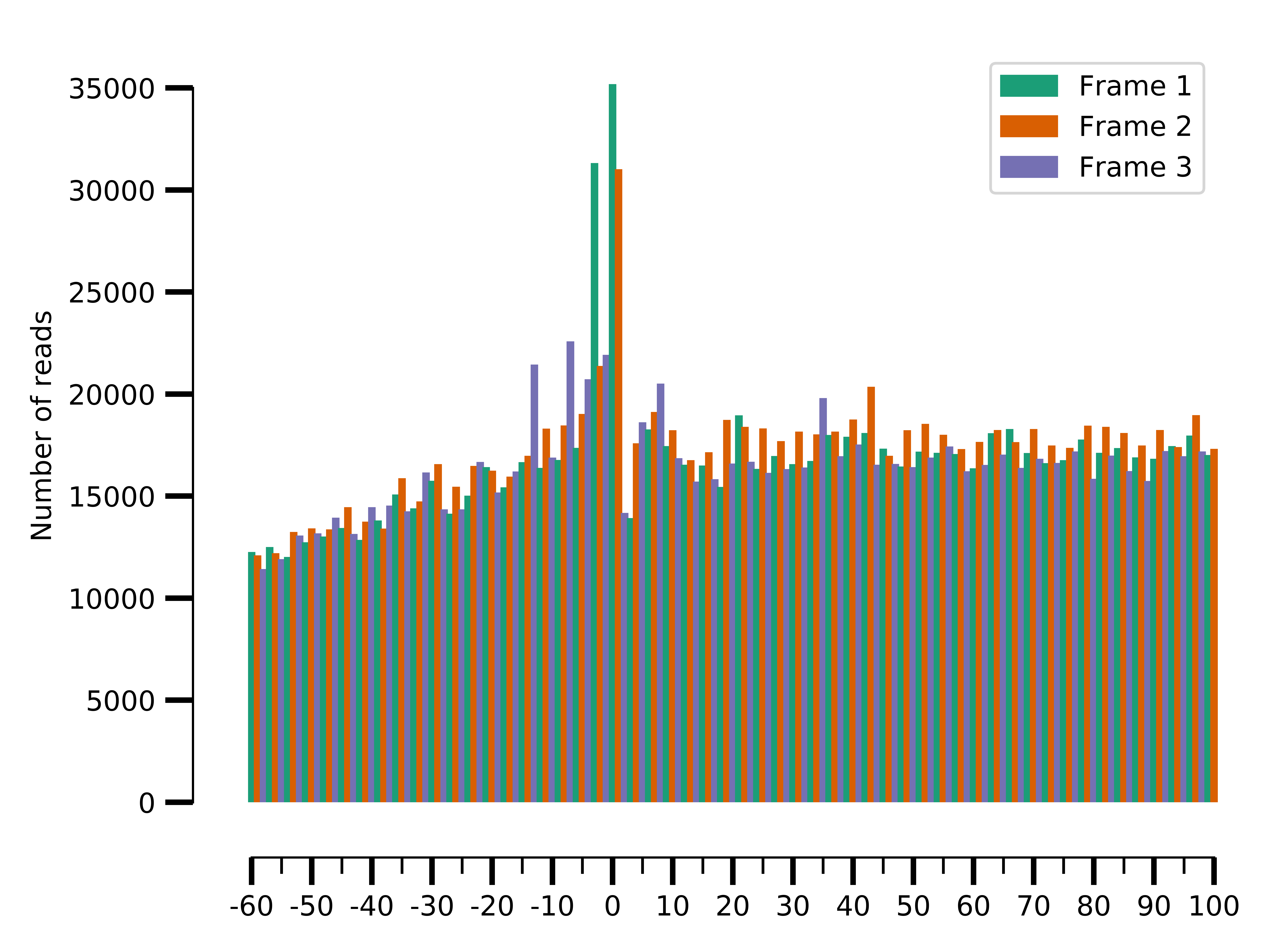
Framewise distribution for SRR5227310
Let’s try another sample: SRR5227306 and compare it with SRR5227310 with respect to distribution of reads.
$ riboraptor uniq-mapping-count --bam data/SRR5227306.bam
10658208
$ riboraptor read-length-dist --bam data/SRR5227306.bam | riboraptor plot-read-dist --saveto SRR5227306.png
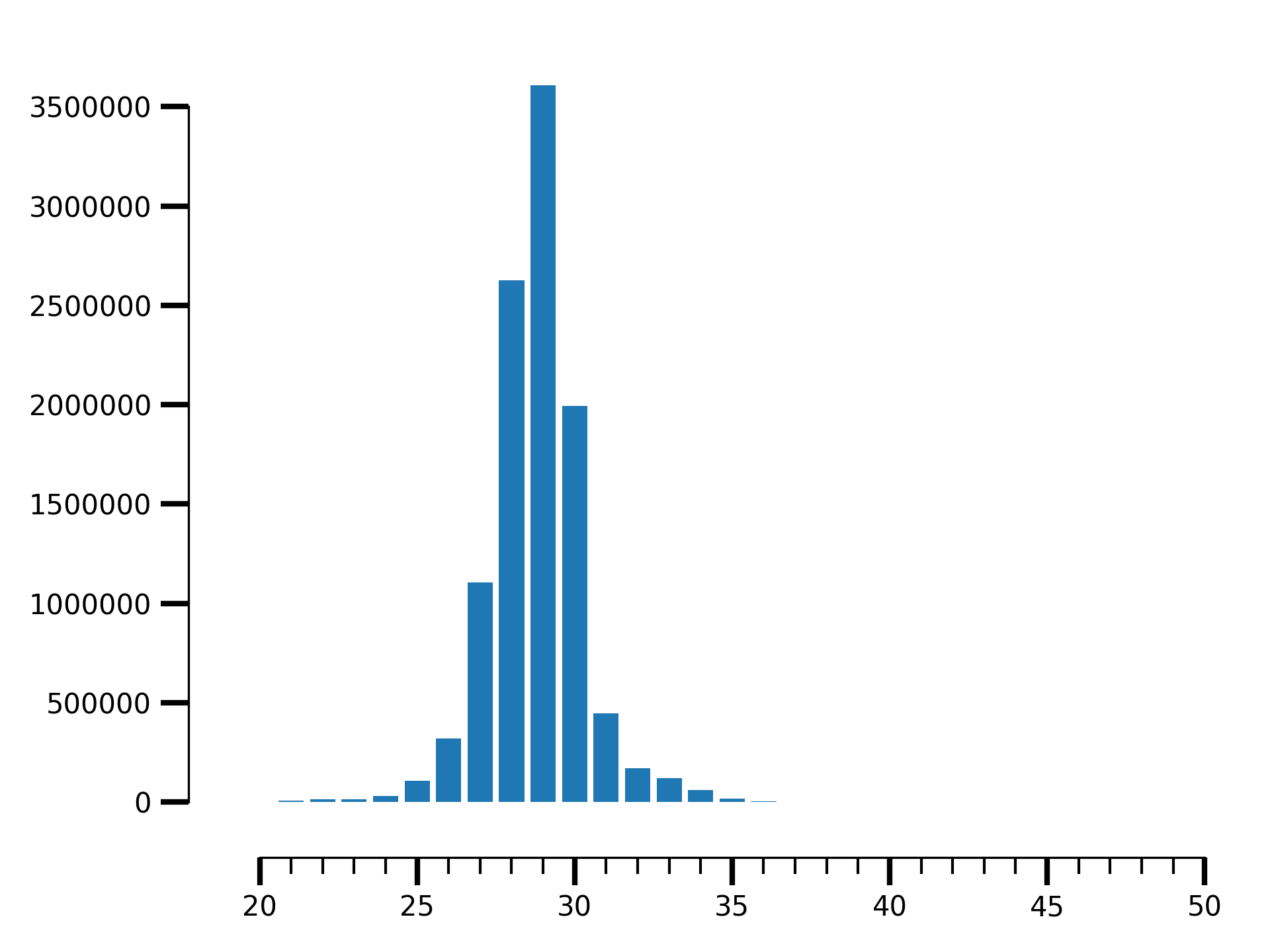
Fragment length distribution for SRR5227306
$ riboraptor read-length-dist --bam data/SRR5227306.bam | riboraptor read-enrichment
(Enrichment: 14.0292145986, pval: 0.135220082438)
As compared to SRR5227310, the enrichment in this case is almost 10 times higher.
$ riboraptor plot-framewise-counts --counts data/SRR5227306.metagene_metagene_raw.pickle\
--saveto data/SRR5227306.framewise.png
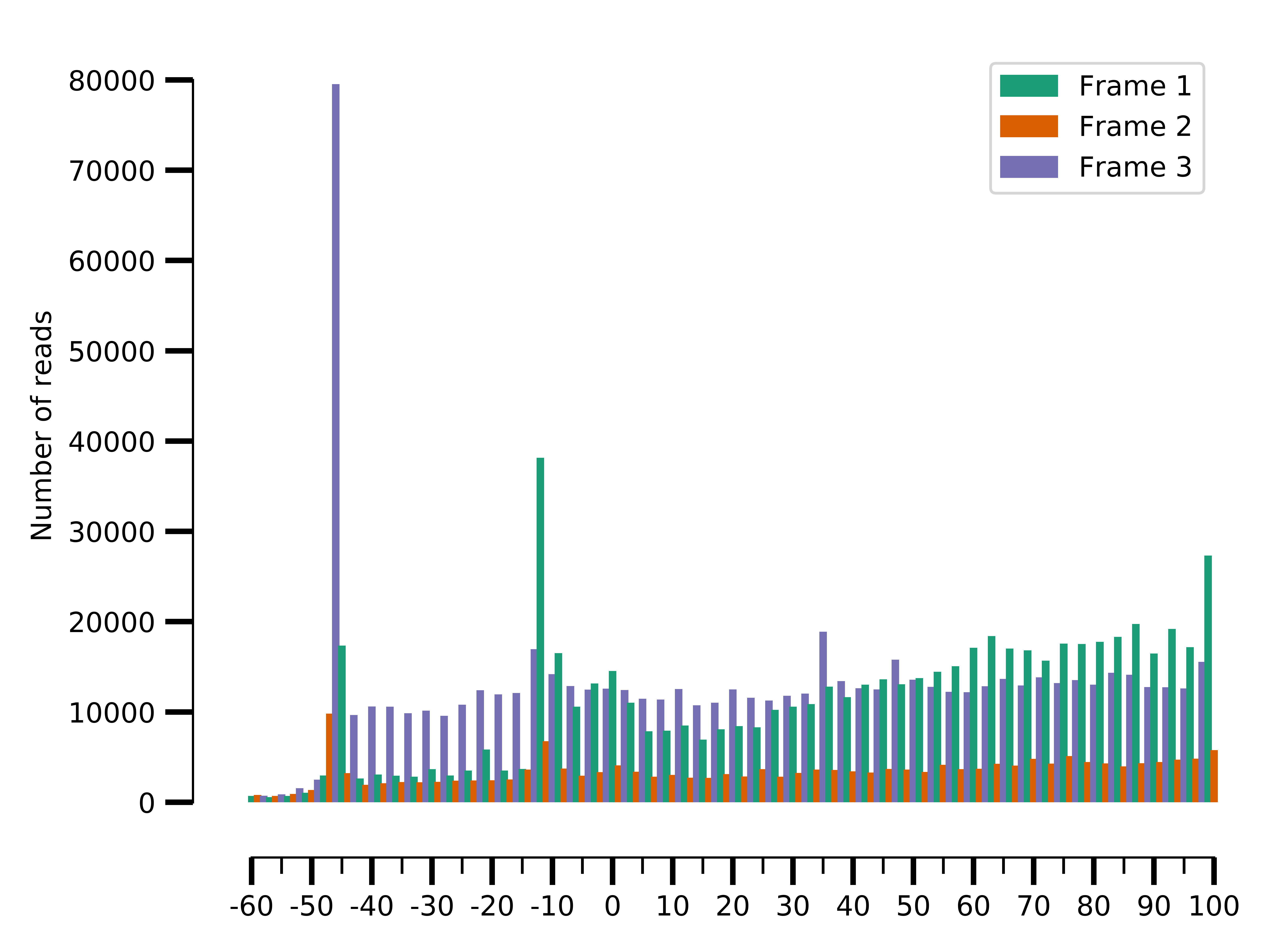
Framewise distribution for SRR5227306
We can see the framewise distribution of reads in SRR5227310 is more or less uniform, but not so in SRR5227306.
$ riboraptor bedgraph-to-bigwig -bg data/SRR5227306.bg -s hg38 -o data/SRR5227306.bw
$ riboraptor metagene-coverage -bw data/SRR5227306.bw \
--region_bed hg38_cds --max-positions 500 \
--prefix data/SRR5227306.metagene --offset 60 --ignore_tx_version
$ riboraptor plot-read-counts \
--counts data/SRR5227306.metagene_metagene_normalized.pickle\
--saveto data/SRR5227306.metagene.png
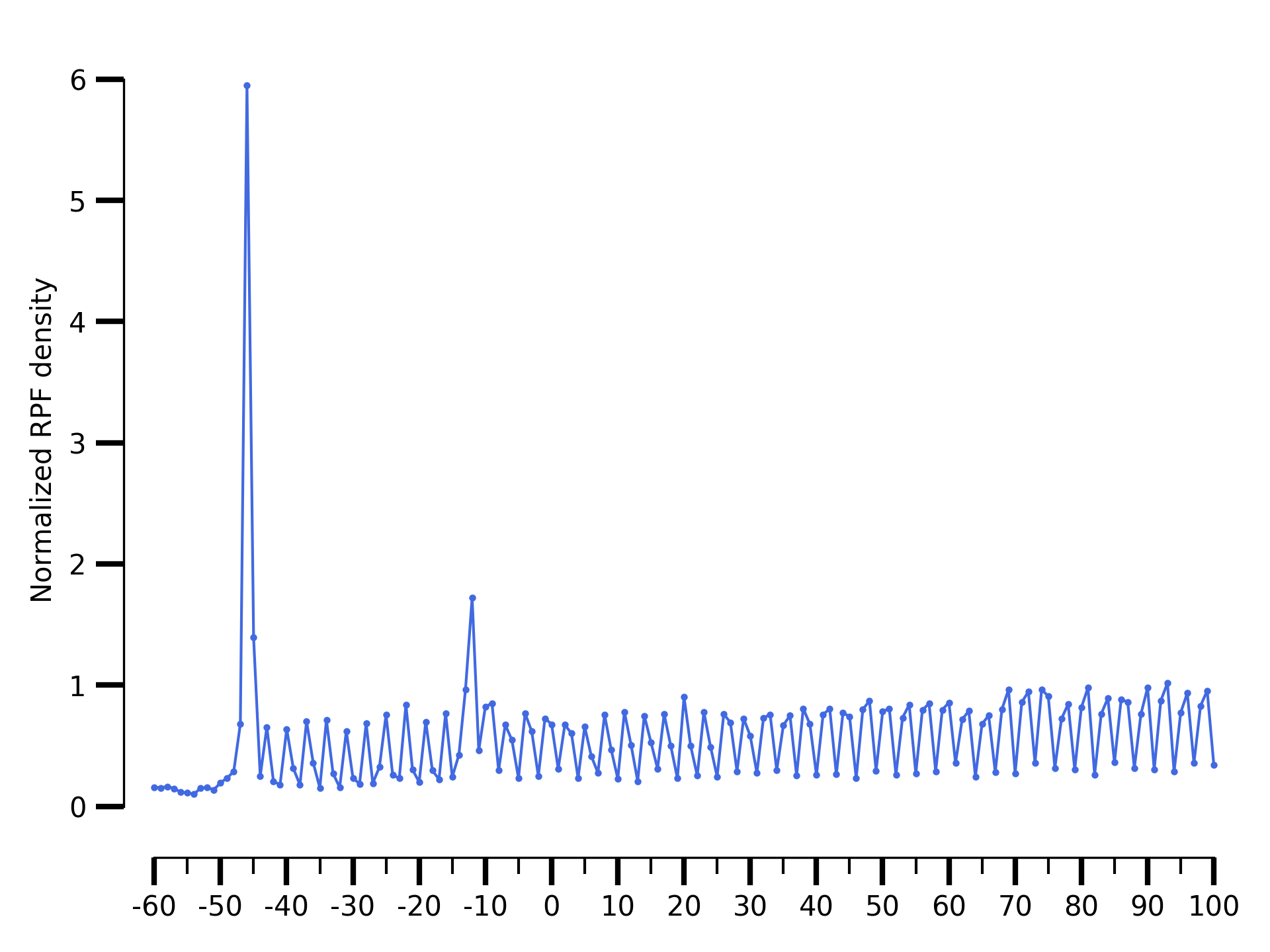
Metagene distribution for SRR5227306
The metagene of a Ribo-seq sample will show periodicity as in the case of SRR5227306 sample. On the other hand a RNA-seq sample like SRR5227310 will tend to have a flat profile.
Distribution of 5’UTR/CDS/3’UTR counts¶
TODO