Welcome to MoCA’s documentation!¶
MoCA: Tool for MOtif Conservation Analysis¶




LICENSE¶
ISC
Installation¶
Current Version¶
0.3.3.dev0
Requirements¶
- bedtools>=2.25.0
- biopython>=1.66
- pandas>=0.18
- scipy>=0.17
- statsmodels>=0.6
- pybigwig>=0.2.8
- seaborn>=0.7.0
- MEME>=4.10.2
NOTE: MoCA also relies on fasta-shuffle-letters that was introduced in MEME 4.11.0 hence if you are using 4.10.2 make sure the fasta-shuffle-letters is the updated one.
For a sample script see travis/install_meme.sh
Using Conda¶
moca is most compatible with the conda environment.
$ conda config --add channels bioconda
$ conda install moca
Using pip¶
$ pip install moca
For development¶
$ git clone https://github.com:saketkc/moca.git
$ cd moca
$ conda env create -f environment.yml python=2.7
$ source activate mocadev
$ python setup.py install
Workflow¶
MoCA makes use of PhyloP/PhastCons/GERP scores to assess the quality of a motif, the hypothesis being a ‘true motif’ would evolve slower as compared to its surrounding(flanking sequences).
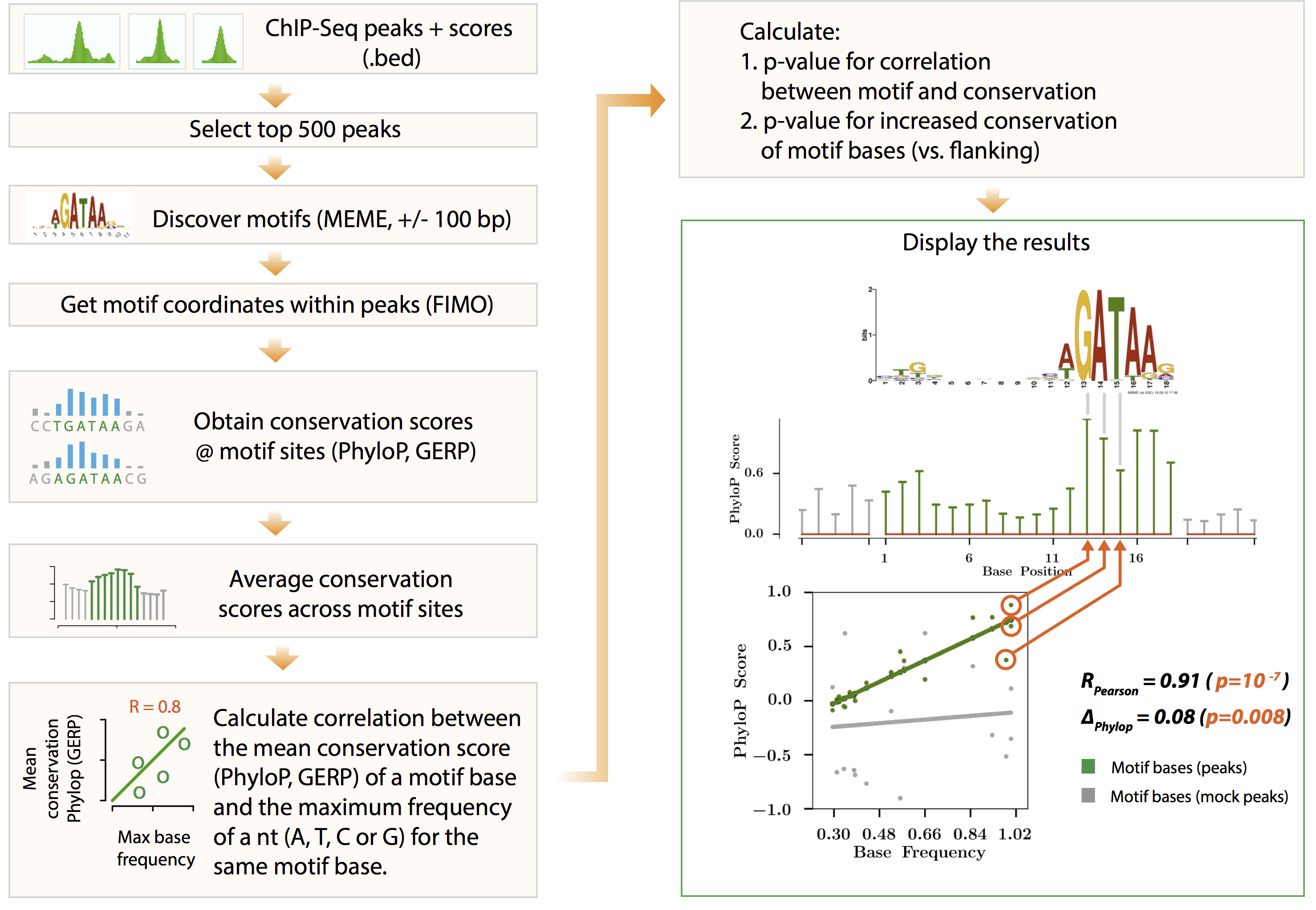
Usage¶
$ moca
Usage: moca [OPTIONS] COMMAND [ARGS]...
moca: Motif Conservation Analysis
Options:
--version Show the version and exit.
--help Show this message and exit.
Commands:
find_motifs Run meme to locate motifs and create...
plot Create stacked conservation plots
Motif analysis using MEME¶
MoCA can perform motif analysis for you given a bedfile containing ChIP-Seq peaks.
Genome builds and MEME binary locations are specified through a configuraton file. A sample configuration file is available: tests/data/application.cfg and should be self-explanatory.
moca find_motifs¶
$ moca find_motifs -h
Usage: moca find_motifs [OPTIONS]
Run meme to locate motifs and create conservation stacked plots
Options:
-i, --bedfile TEXT Bed file input [required]
-o, --oc TEXT Output Directory [required]
-c, --configuration TEXT Configuration file [required]
--slop-length INTEGER Flanking sequence length [required]
--flank-motif INTEGER Length of sequence flanking motif [required]
--n-motif INTEGER Number of motifs
-t, --cores INTEGER Number of parallel MEME jobs [required]
-g, -gb, --genome-build TEXT Key denoting genome build to use in
configuration file [required]
--show-progress Print progress
-h, --help Show this message and exit.
moca plot¶
$ moca plot -h
Usage: moca plot [OPTIONS]
Create stacked conservation plots
Options:
--meme-dir, --meme_dir TEXT MEME output directory [required]
--centrimo-dir, --centrimo_dir TEXT
Centrimo output directory [required]
--fimo-dir-sample, --fimo_dir_sample TEXT
Sample fimo.txt [required]
--fimo-dir-control, --fimo_dir_control TEXT
Control fimo.txt [required]
--name TEXT Plot title
--flank-motif INTEGER Length of sequence flanking motif
[required]
--motif INTEGER Motif number
-o, --oc TEXT Output Directory [required]
-c, --configuration TEXT Configuration file [required]
--show-progress Print progress
-g, -gb, --genome-build TEXT Key denoting genome build to use in
configuration file [required]
-h, --help Show this message and exit.
Example¶
Most users will require using the command line version only:
$ moca find_motifs -i encode_test_data/ENCFF002DAR.bed\
-c tests/data/application.cfg -g hg19 --show-progress
Creating plots if you already have run MEME and Centrimo:
$ moca plot -c tests/data/application.cfg -g hg19\
--meme-dir moca_output/meme_out\
--centrimo-dir moca_output/centrimo_out\
--fimo-dir-sample moca_output/meme_out/fimo_out_1\
--fimo-dir-control moca_output/meme_out/fimo_random_1\
--name ENCODEID
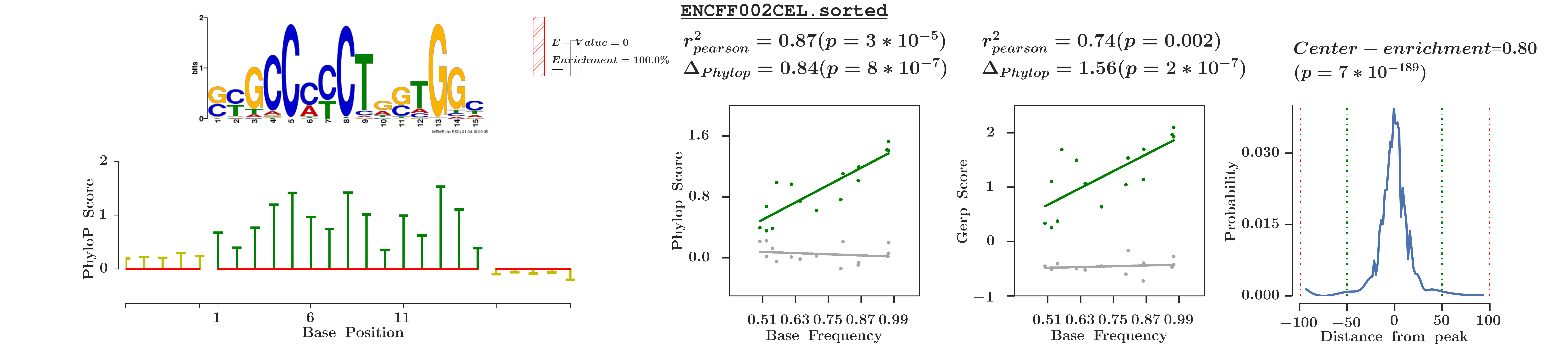
There is also a structured API available, however it might be missing examples and documentation at places.
API Documentation¶
Credits¶
This package was created with Cookiecutter and the audreyr/cookiecutter-pypackage project template.
moca¶
Contents: